Biological Entity Dictionary (BED): exploring and converting identifiers of biological entities such as genes, transcripts or peptides
Source:vignettes/BED.Rmd
BED.Rmd
Introduction
This Biological Entity Dictionary (BED) has been developed to address three main challenges. The first one is related to the completeness of identifier mappings. Indeed, direct mapping information provided by the different systems are not always complete and can be enriched by mappings provided by other resources. More interestingly, direct mappings not identified by any of these resources can be indirectly inferred by using mappings to a third reference. For example, many human Ensembl gene ID are not directly mapped to any Entrez gene ID but such mappings can be inferred using respective mappings to HGNC ID. The second challenge is related to the mapping of deprecated identifiers. Indeed, entity identifiers can change from one resource release to another. The identifier history is provided by some resources, such as Ensembl or the NCBI, but it is generally not used by mapping tools. The third challenge is related to the automation of the mapping process according to the relationships between the biological entities of interest. Indeed, mapping between gene and protein ID scopes should not be done the same way than between two scopes regarding gene ID. Also, converting identifiers from different organisms should be possible using gene orthologs information.
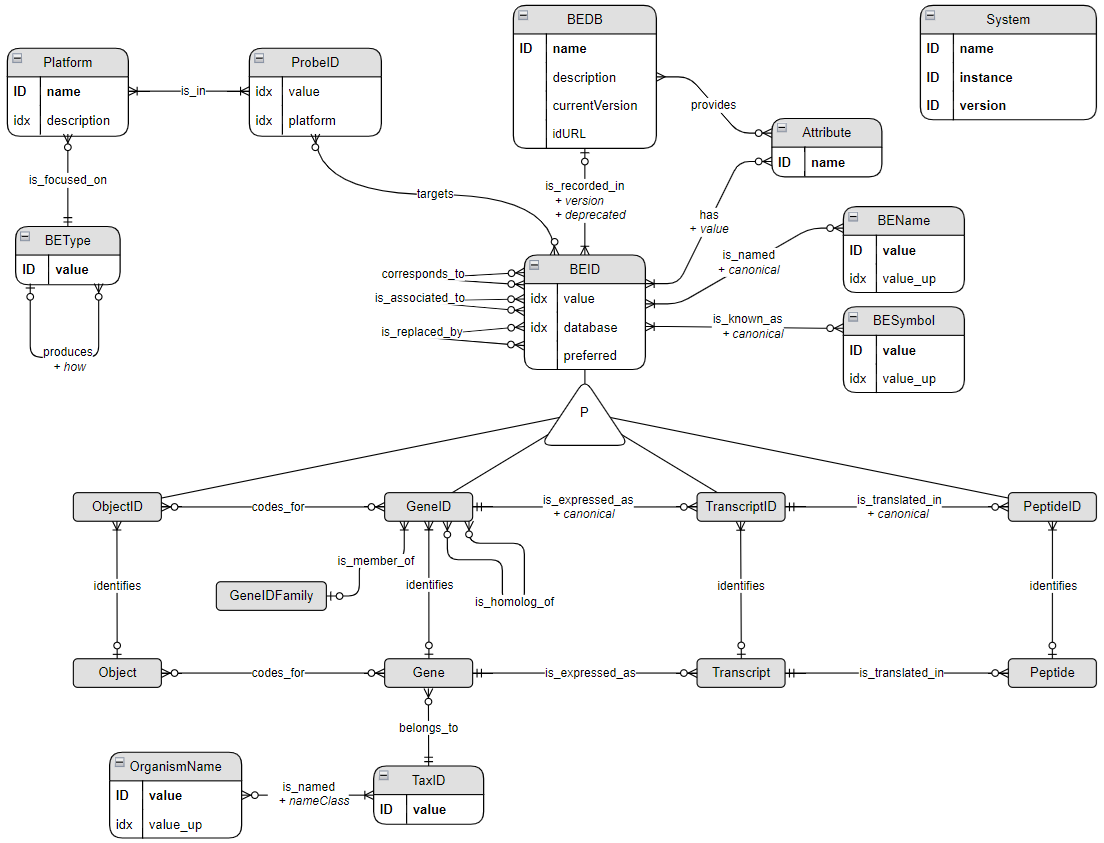
This document shows how to use the BED (Biological Entity Dictionary) R package to get and explore mapping between identifiers of biological entities (BE). This package provides a way to connect to a BED Neo4j database in which the relationships between the identifiers from different sources are recorded.
Citing BED
This package and the underlying research has been published in this peer reviewed article:
Installation
Dependencies
This BED package depends on the following packages available in the CRAN repository:
- neo2R
- visNetwork
- dplyr
- readr
- stringr
- utils
- shiny
- DT
- miniUI
- rstudioapi
All these packages must be installed before installing BED.
Installation from github
devtools::install_github("patzaw/BED")Possible issue when updating from releases <= 1.3.0
If you get an error like the following…
Error: package or namespace load failed for ‘BED’:
.onLoad failed in loadNamespace() for 'BED', details:
call: connections[[connection]][["cache"]]
error: subscript out of bounds… remove the BED folder located here:
file.exists(file.path(Sys.getenv("HOME"), "R", "BED"))Connection
Before using BED, the connection needs to be established with the
underlying Neo4j DB. url, username and
password should be adapted.
connectToBed(url="localhost:5454", remember=FALSE, useCache=FALSE)The remember parameter can be set to TRUE
in order to save connection information that will be automatically used
the next time the connectToBed() function is called. By
default, this parameter is set to FALSE to comply with CRAN
policies. Saved connection can be managed with the
lsBedConnections() and the
forgetBedConnection() functions.
The useCache parameter is by default set to
FALSE to comply with CRAN policies. However, it is
recommended to set it to TRUE to improve the speed of
recurrent queries: the results of some large queries are saved locally
in a file.
The connection can be checked the following way.
checkBedConn(verbose=TRUE)## https://genodesy.org/BED## BED## Genodesy-Human## 2026.06.27## Cache ON## [1] TRUE
## attr(,"dbVersion")
## name instance version
## 1 BED Genodesy-Human 2026.06.27If the verbose parameter is set to TRUE, the URL and the
content version are displayed as messages.
The following function list saved connections.
The connection param of the connectToBed
function can be used to connect to a saved connection other than the
last one.
Direct calls
Cypher queries can be run directly on the Neo4j database using the
cypher function from the neo2R package
through the bedCall function.
results <- bedCall(
cypher,
query=prepCql(
'MATCH (n:BEID)',
'WHERE n.value IN $values',
'RETURN DISTINCT n.value AS value, labels(n), n.database'
),
parameters=list(values=c("10", "100"))
)
results## value labels(n) n.database
## 1 10 BEID || GeneID EntrezGene
## 2 100 BEID || GeneID EntrezGene
## 3 100 BEID || GeneID HGNCFeeding the database
Many functions are provided within the package to build your own BED database instance. These functions are not exported in order to avoid their use when interacting with BED normally. Information about how to get an instance of the BED neo4j database is provided here:
- https://github.com/patzaw/BED#bed-database-instance-available-as-a-docker-image
- https://github.com/patzaw/BED#build-a-bed-database-instance
It can be adapted to user needs.
Caching
This part is relevant if the useCache parameter is set
to TRUE when calling connectToBed().
Functions of the BED package used to retrieve thousands of identifiers can take some time (generally a few seconds) before returning a result. Thus for this kind of query, the query is run for all the relevant ID in the DB and thanks to a cache system implemented in the package same queries with different filters should be much faster the following times.
By default the cache is flushed when the system detect
inconsistencies with the BED database. However, it can also be manualy
flushed if needed using the clearBedCache() function.
Queries already in cache can be listed using the
lsBedCache() function which also return the occupied disk
space.
Exploring available data
Biological entities
BED is organized around the central concept of Biological Entity (BE). All supported types of BE can be listed.
listBe()## [1] "Gene" "Transcript" "Peptide" "Object"These BE are organized according to how they are related to each other. For example a Gene is_expressed_as a Transcript. This organization allows to find the first upstream BE common to a set of BE.
firstCommonUpstreamBe(c("Object", "Transcript"))## [1] "Gene"
firstCommonUpstreamBe(c("Peptide", "Transcript"))## [1] "Transcript"Organisms
Several organims can be supported by the BED underlying database. They can be listed the following way.
## [1] "Danio rerio" "Homo sapiens" "Sus scrofa"
## [4] "Mus musculus" "Rattus norvegicus"Common names are also supported and the corresponding taxonomic identifiers can be retrieved. Conversely the organism names corresponding to a taxonomic ID can be listed.
getOrgNames(getTaxId("human"))## taxID name nameClass
## 1 9606 Homo sapiens Linnaeus, 1758 authority
## 2 9606 human genbank common name
## 3 9606 Homo sapiens scientific nameIdentifiers of biological entities
The main aim of BED is to allow the mapping of identifiers from different sources such as Ensembl or Entrez. Supported sources can be listed the following way for each supported organism.
listBeIdSources(be="Transcript", organism="human")## database nbBe nbId be
## 1 BEDTech_transcript 54696 54696 Transcript
## 2 RefSeq 270254 279191 Transcript
## 3 Ens_transcript 666691 681719 TranscriptThe database gathering the largest number of BE of specific type can also be identified.
largestBeSource(be="Transcript", organism="human", restricted=TRUE)## [1] "Ens_transcript"Finally, the getAllBeIdSources() function returns all
the source databases of BE identifiers whatever the BE type.
Experimental platforms and probes
BED also supports experimental platforms and provides mapping betweens probes and BE identifiers (BEID).
The supported platforms can be listed the following way. The
getTargetedBe() function returns the type of BE on which a
specific platform focus.
## name description focus
## GPL6101 GPL6101 Illumina ratRef-12 v1.0 expression beadchip Gene
## GPL6887 GPL6887 Illumina MouseWG-6 v2.0 expression beadchip Gene
## GPL6947 GPL6947 Illumina HumanHT-12 V3.0 expression beadchip Gene
## GPL10558 GPL10558 Illumina HumanHT-12 V4.0 expression beadchip Gene
## GPL1355 GPL1355 [Rat230_2] Affymetrix Rat Genome 230 2.0 Array Gene
## GPL1261 GPL1261 [Mouse430_2] Affymetrix Mouse Genome 430 2.0 Array Gene
getTargetedBe("GPL570")## [1] "Gene"Managing identifiers
Retrieving all identifiers from a source
All identifiers of an organism BEs from one source can be retrieved.
## [1] 216661 5
head(beids)## id preferred Gene db.version db.deprecated
## 1 4535 FALSE 974767 20260627 FALSE
## 2 4536 FALSE 974771 20260627 FALSE
## 3 4512 FALSE 974777 20260627 FALSE
## 4 4513 FALSE 974780 20260627 FALSE
## 5 4509 FALSE 974782 20260627 FALSE
## 6 4508 FALSE 974783 20260627 FALSEThe first column, id, corresponds to the identifiers of the BE in the source. The column named according to the BE type (in this case Gene) corresponds to the internal identifier of the related BE. BE CAREFUL, THIS INTERNAL ID IS NOT STABLE AND CANNOT BE USED AS A REFERENCE. This internal identifier is useful to identify BEIDS corresponding to the same BE. The following code can be used to have an overview of such redundancy.
##
## 1 2 3 4 5 6 7 8 9 10 11
## 178540 9837 3186 1065 405 161 92 47 25 11 10
## 12 13 14 16 31
## 6 2 2 1 1In the example above we can see that most of Gene BE are identified
by only one EntrezGene ID. However many of them are identified by two or
more ID; 33 BE are even identified by 10 or more EntrezGeneID. In this
case, most of these redundancies come from ID history extracted from
Entrez. Legacy ID can be excluded from the retrieved ID by setting the
restricted parameter to TRUE.
## [1] 193821 5The same code as above can be used to identify remaining redundancies.
##
## 1 2 3 4
## 192988 378 23 2In the example above we can see that allmost all Gene BE are identified by only one EntrezGene ID. However some of them are identified by two or more ID. This result comes from how the BED database is constructed according to the ID mapping provided by the different source databases. The graph below shows how the mapping was done for such a BE with redundant EntrezGene IDs.
This issue has been mainly solved by not taking into account
ambigous mappings between NCBI Entrez gene identifiers and Ensembl gene
identifier provided by Ensembl. It has been achieved using the
cleanDubiousXRef() function from the 2019.10.11 version of
the BED-UCB-Human database.
The way the ID correspondances are reported in the different source databases leads to this mapping ambiguity which has to be taken into account when comparing identifiers from different databases.
The getBeIds() returns other columns providing
additional information about the id. The same function can be
used to retrieved symbols or probe identifiers.
Preferred identifier
The BED database is constructed according to the relationships
between identifiers provided by the different sources. Biological
entities (BE) are identified as clusters of identifiers which correspond
to each other directly or indirectly (corresponds_to
relationship). Because of this design a BE can be identified by multiple
identifiers (BEID) from the same database as shown above. These BEID are
often related to alternate version of an entity.
For example, Ensembl provides different version (alternative sequences) of some chromosomes parts. And genes are also annotated on these alternative sequences. In Uniprot some unreviewed identifiers can correspond to reviewed proteins.
When available such kind of information is associated to an
Attribute node through a has relationship
providing the value of the attribute for the BEID. This information can
also be used to define if a BEID is a preferred identifier for
a BE.
The example below shows the case of the MAPT gene annotated on different version of human chromosome 17.
Checking identifiers
The origin of identifiers can be guessed as following.
oriId <- c(
"17237", "105886298", "76429", "80985", "230514", "66459",
"93696", "72514", "20352", "13347", "100462961", "100043346",
"12400", "106582", "19062", "245607", "79196", "16878", "320727",
"230649", "66880", "66245", "103742", "320145", "140795"
)
idOrigin <- guessIdScope(oriId)
print(idOrigin$be)## [1] "Gene"
print(idOrigin$source)## [1] "EntrezGene"
print(idOrigin$organism)## [1] "Mus musculus"The best guess is returned as a list but other possible origins are listed in the details attribute.
## be source organism nb proportion
## 1 Gene EntrezGene Mus musculus 25 1.00
## 2 Gene HGNC Homo sapiens 3 0.12
## 3 Gene MGI Mus musculus 2 0.08If the origin of identifiers is already known, it can also be tested.
checkBeIds(ids=oriId, be="Gene", source="EntrezGene", organism="mouse")
checkBeIds(ids=oriId, be="Gene", source="HGNC", organism="human")## Warning in checkBeIds(ids = oriId, be = "Gene", source = "HGNC", organism =
## "human"): Could not find 22 IDs among 25!Identifier annotation
Identifiers can be annotated with symbols and names according to
available information. The following code returns the most relevant
symbol and the most relevant name for each ID. Source URL can also be
generated with the getBeIdURL() function.
toShow <- getBeIdDescription(
ids=oriId, be="Gene", source="EntrezGene", organism="mouse"
)
toShow$id <- paste0(
sprintf(
'<a href="%s" target="_blank">',
getBeIdURL(toShow$id, "EntrezGene")
),
toShow$id,
'<a>'
)
kable(toShow, escape=FALSE, row.names=FALSE)| id | symbol | name | preferred | db.version | db.deprecated |
|---|---|---|---|---|---|
| 17237 | Mgrn1 | mahogunin, ring finger 1 | TRUE | 20260627 | FALSE |
| 105886298 | Cmc4 | C-X9-C motif containing 4 | TRUE | 20260627 | FALSE |
| 76429 | Lhpp | phospholysine phosphohistidine inorganic pyrophosphate phosphatase | TRUE | 20260627 | FALSE |
| 80985 | Trim44 | tripartite motif-containing 44 | TRUE | 20260627 | FALSE |
| 230514 | Leprot | leptin receptor overlapping transcript | TRUE | 20260627 | FALSE |
| 66459 | Pyurf | Pigy upstream reading frame | TRUE | 20260627 | FALSE |
| 93696 | Chrac1 | chromatin accessibility complex 1 | TRUE | 20260627 | FALSE |
| 72514 | Fgfbp3 | fibroblast growth factor binding protein 3 | TRUE | 20260627 | FALSE |
| 20352 | Sema4b | sema domain, immunoglobulin domain (Ig), transmembrane domain (TM) and short cytoplasmic domain, (semaphorin) 4B | TRUE | 20260627 | FALSE |
| 13347 | Dffa | DNA fragmentation factor, alpha subunit | TRUE | 20260627 | FALSE |
| 100462961 | Gm16149 | predicted gene 16149 | TRUE | 20260627 | FALSE |
| 100043346 | Rpl10-ps3 | ribosomal protein L10, pseudogene 3 | TRUE | 20260627 | FALSE |
| 12400 | Cbfb | core binding factor beta | TRUE | 20260627 | FALSE |
| 106582 | Nrm | nurim (nuclear envelope membrane protein) | TRUE | 20260627 | FALSE |
| 19062 | Inpp5k | inositol polyphosphate 5-phosphatase K | TRUE | 20260627 | FALSE |
| 245607 | Gprasp2 | G protein-coupled receptor associated sorting protein 2 | TRUE | 20260627 | FALSE |
| 79196 | Osbpl5 | oxysterol binding protein-like 5 | TRUE | 20260627 | FALSE |
| 16878 | Lif | leukemia inhibitory factor | TRUE | 20260627 | FALSE |
| 320727 | Ipo8 | importin 8 | TRUE | 20260627 | FALSE |
| 230649 | Atpaf1 | ATP synthase mitochondrial F1 complex assembly factor 1 | TRUE | 20260627 | FALSE |
| 66880 | Rsrc1 | arginine/serine-rich coiled-coil 1 | TRUE | 20260627 | FALSE |
| 66245 | Hspbp1 | HSPA (heat shock 70kDa) binding protein, cytoplasmic cochaperone 1 | TRUE | 20260627 | FALSE |
| 103742 | Mien1 | migration and invasion enhancer 1 | TRUE | 20260627 | FALSE |
| 320145 | Sp8 | trans-acting transcription factor 8 | TRUE | 20260627 | FALSE |
| 140795 | P2ry14 | purinergic receptor P2Y, G-protein coupled, 14 | TRUE | 20260627 | FALSE |
All possible symbols and all possible names for each ID can also be retrieved using the following functions.
res <- getBeIdSymbols(
ids=oriId, be="Gene", source="EntrezGene", organism="mouse",
restricted=FALSE
)
head(res)## id symbol canonical direct preferred entity
## 1 66459 Prey FALSE TRUE TRUE 4601575
## 2 66459 2610022G08Rik FALSE TRUE TRUE 4601575
## 3 66459 Pyurf TRUE TRUE TRUE 4601575
## 4 100043346 Gm10041 FALSE TRUE TRUE 10838497
## 5 100043346 Rpl10-ps3 TRUE TRUE TRUE 10838497
## 6 16878 Lif TRUE TRUE TRUE 4654819
res <- getBeIdNames(
ids=oriId, be="Gene", source="EntrezGene", organism="mouse",
restricted=FALSE
)
head(res)## id name canonical direct preferred
## 1 66459 pre-Y homolog FALSE TRUE TRUE
## 2 66459 protein preY, mitochondrial FALSE TRUE TRUE
## 3 66459 Pigy upstream reading frame TRUE TRUE TRUE
## 4 100043346 ribosomal protein L20, pseudogene 3 FALSE TRUE TRUE
## 5 100043346 ribosomal protein L10 pseudogene FALSE TRUE TRUE
## 6 100043346 predicted pseudogene 10041 FALSE TRUE TRUE
## entity
## 1 4601575
## 2 4601575
## 3 4601575
## 4 10838497
## 5 10838497
## 6 10838497Also probes and some biological entities do not have directly associated symbols or names. These elements can also be annotated according to information related to relevant genes.
someProbes <- c(
"238834_at", "1569297_at", "213021_at", "225480_at",
"216016_at", "35685_at", "217969_at", "211359_s_at"
)
toShow <- getGeneDescription(
ids=someProbes, be="Probe", source="GPL570", organism="human"
)
kable(toShow, escape=FALSE, row.names=FALSE)| id | EntrezGene | symbol | name |
|---|---|---|---|
| 238834_at | 91807 | MYLK3 | myosin light chain kinase 3 |
| 1569297_at | 731779 | LINC01300 | long independently transcribed non-coding RNA 1300 |
| 213021_at | 9527 | GOSR1 | golgi SNAP receptor complex member 1 |
| 225480_at | 127687 | C1orf122 | chromosome 1 open reading frame 122 |
| 216016_at | 114548 | NLRP3 | NLR family pyrin domain containing 3 |
| 35685_at | 6015 | RING1 | ring finger protein 1 |
| 217969_at | 738 | VPS51 | VPS51 subunit of GARP complex |
| 211359_s_at | 4988 | OPRM1 | opioid receptor mu 1 |
Products of molecular biology processes
The BED data model has beeing built to fulfill molecular biology processes:
- is_expressed_as relationships correspond to the transcription process.
- is_translated_in relationships correspond to the translation process.
- codes_for is a fuzzy relationship allowing the mapping of genes on object not necessary corresonpding to the same kind of biological molecule.
These processes are described in different databases with different level of granularity. For exemple, Ensembl provides possible transcripts for each gene specifying which one of them is canonical.
The following functions are used to retrieve direct products or direct origins of molecular biology processes.
getDirectProduct("ENSG00000145335", process="is_expressed_as")## origin osource product psource canonical
## 1 ENSG00000145335 Ens_gene ENST00000889660 Ens_transcript FALSE
## 2 ENSG00000145335 Ens_gene ENST00000965189 Ens_transcript FALSE
## 3 ENSG00000145335 Ens_gene ENST00000394991 Ens_transcript TRUE
## 4 ENSG00000145335 Ens_gene ENST00000673902 Ens_transcript FALSE
## 5 ENSG00000145335 Ens_gene ENST00001098870 Ens_transcript FALSE
## 6 ENSG00000145335 Ens_gene ENST00000965188 Ens_transcript FALSE
## 7 ENSG00000145335 Ens_gene ENST00001098868 Ens_transcript FALSE
## 8 ENSG00000145335 Ens_gene ENST00001098871 Ens_transcript FALSE
## 9 ENSG00000145335 Ens_gene ENST00000611107 Ens_transcript FALSE
## 10 ENSG00000145335 Ens_gene ENST00000508895 Ens_transcript FALSE
## 11 ENSG00000145335 Ens_gene ENST00001098866 Ens_transcript FALSE
## 12 ENSG00000145335 Ens_gene ENST00000889662 Ens_transcript FALSE
## 13 ENSG00000145335 Ens_gene ENST00000506691 Ens_transcript FALSE
## 14 ENSG00000145335 Ens_gene ENST00000505199 Ens_transcript FALSE
## 15 ENSG00000145335 Ens_gene ENST00000394986 Ens_transcript FALSE
## 16 ENSG00000145335 Ens_gene ENST00000673766 Ens_transcript FALSE
## 17 ENSG00000145335 Ens_gene ENST00000889658 Ens_transcript FALSE
## 18 ENSG00000145335 Ens_gene ENST00000336904 Ens_transcript FALSE
## 19 ENSG00000145335 Ens_gene ENST00000889659 Ens_transcript FALSE
## 20 ENSG00000145335 Ens_gene ENST00000912364 Ens_transcript FALSE
## 21 ENSG00000145335 Ens_gene ENST00000674129 Ens_transcript FALSE
## 22 ENSG00000145335 Ens_gene ENST00000965187 Ens_transcript FALSE
## 23 ENSG00000145335 Ens_gene ENST00000965192 Ens_transcript FALSE
## 24 ENSG00000145335 Ens_gene ENST00000673718 Ens_transcript FALSE
## 25 ENSG00000145335 Ens_gene ENST00000345009 Ens_transcript FALSE
## 26 ENSG00000145335 Ens_gene ENST00000889663 Ens_transcript FALSE
## 27 ENSG00000145335 Ens_gene ENST00000506244 Ens_transcript FALSE
## 28 ENSG00000145335 Ens_gene ENST00001013821 Ens_transcript FALSE
## 29 ENSG00000145335 Ens_gene ENST00000502987 Ens_transcript FALSE
## 30 ENSG00000145335 Ens_gene ENST00000618500 Ens_transcript FALSE
## 31 ENSG00000145335 Ens_gene ENST00000965190 Ens_transcript FALSE
## 32 ENSG00000145335 Ens_gene ENST00001098869 Ens_transcript FALSE
## 33 ENSG00000145335 Ens_gene ENST00000394989 Ens_transcript FALSE
## 34 ENSG00000145335 Ens_gene ENST00000912365 Ens_transcript FALSE
## 35 ENSG00000145335 Ens_gene ENST00000420646 Ens_transcript FALSE
## 36 ENSG00000145335 Ens_gene ENST00000965186 Ens_transcript FALSE
## 37 ENSG00000145335 Ens_gene ENST00000965191 Ens_transcript FALSE
## 38 ENSG00000145335 Ens_gene ENST00001133508 Ens_transcript FALSE
## 39 ENSG00000145335 Ens_gene ENST00000889657 Ens_transcript FALSE
## 40 ENSG00000145335 Ens_gene ENST00001098867 Ens_transcript FALSE
getDirectProduct("ENST00000336904", process="is_translated_in")## origin osource product psource canonical
## 1 ENST00000336904 Ens_transcript ENSP00000338345 Ens_translation TRUE
getDirectOrigin("NM_001146055", process="is_expressed_as")## origin osource product psource canonical
## 1 6622 EntrezGene NM_001146055 RefSeq FALSEConverting identifiers
Same entity and same organism: from one source to another
res <- convBeIds(
ids=oriId,
from="Gene",
from.source="EntrezGene",
from.org="mouse",
to.source="Ens_gene",
restricted=TRUE,
prefFilter=TRUE
)
head(res)## from to to.preferred to.entity
## 1 66459 ENSMUSG00000043162 TRUE 4601575
## 2 72514 ENSMUSG00000047632 TRUE 4621515
## 3 105886298 ENSMUSG00000090110 TRUE 4646923
## 4 245607 ENSMUSG00000072966 TRUE 4648306
## 5 320145 ENSMUSG00000048562 TRUE 4648473
## 6 13347 ENSMUSG00000028974 TRUE 4649175Same organism: from one entity to another
res <- convBeIds(
ids=oriId,
from="Gene",
from.source="EntrezGene",
from.org="mouse",
to="Peptide",
to.source="Ens_translation",
restricted=TRUE,
prefFilter=TRUE
)
head(res)## from to to.preferred to.entity
## 1 66459 ENSMUSP00000050958 TRUE 6870080
## 4 72514 ENSMUSP00000059682 TRUE 6877698
## 2 72514 ENSMUSP00000159137 TRUE 6877698
## 6 72514 ENSMUSP00000199874 TRUE 6877703
## 11 105886298 ENSMUSP00000033543 TRUE 6915056
## 15 105886298 ENSMUSP00000112753 TRUE 6915056From one organism to another
res <- convBeIds(
ids=oriId,
from="Gene",
from.source="EntrezGene",
from.org="mouse",
to="Peptide",
to.source="Ens_translation",
to.org="human",
restricted=TRUE,
prefFilter=TRUE
)
head(res)## from to to.preferred to.entity
## 179 106582 ENSP00000397892 TRUE 3925123
## 632 20352 ENSP00000453272 TRUE 3942040
## 633 20352 ENSP00000452957 TRUE 3942047
## 634 20352 ENSP00000453951 TRUE 3942051
## 635 20352 ENSP00000453705 TRUE 3942209
## 636 20352 ENSP00000453563 TRUE 3942217Converting lists of identifiers
List of identifiers can be converted the following way. Only converted IDs are returned in this case.
humanEnsPeptides <- convBeIdLists(
idList=list(a=oriId[1:5], b=oriId[-c(1:5)]),
from="Gene",
from.source="EntrezGene",
from.org="mouse",
to="Peptide",
to.source="Ens_translation",
to.org="human",
restricted=TRUE,
prefFilter=TRUE
)
unlist(lapply(humanEnsPeptides, length))## a b
## 114 340
lapply(humanEnsPeptides, head)## $a
## [1] "ENSP00000468171" "ENSP00000467414" "ENSP00000464811" "ENSP00000465249"
## [5] "ENSP00000665118" "ENSP00000665119"
##
## $b
## [1] "ENSP00000397892" "ENSP00000453272" "ENSP00000452957" "ENSP00000453951"
## [5] "ENSP00000453705" "ENSP00000453563"BEIDList
BEIDList objects are used to manage lists of BEID with
an attached explicit scope, and metadata provided in a data frame. The
focusOnScope() function is used to easily convert such
object to another scope. For example, in the code below, Entrez gene
identifiers are converted in Ensembl identifiers.
entrezGenes <- BEIDList(
list(a=oriId[1:5], b=oriId[-c(1:5)]),
scope=list(be="Gene", source="EntrezGene", organism="Mus musculus"),
metadata=data.frame(
.lname=c("a", "b"),
description=c("Identifiers in a", "Identifiers in b"),
stringsAsFactors=FALSE
)
)
entrezGenes## BEIDList of 2 elements gathering 25 BEIDs in total
## - Scope: be="Gene", source="EntrezGene", organism="Mus musculus"
## - Metadata fields: ".lname", "description"
entrezGenes$a## [1] "17237" "105886298" "76429" "80985" "230514"
ensemblGenes <- focusOnScope(entrezGenes, source="Ens_gene")
ensemblGenes$a## [1] "ENSMUSG00000090110" "ENSMUSG00000022517" "ENSMUSG00000035212"
## [4] "ENSMUSG00000027189" "ENSMUSG00000030946"Converting data frames
IDs in data frames can also be converted.
toConv <- data.frame(a=1:25, b=runif(25))
rownames(toConv) <- oriId
res <- convDfBeIds(
df=toConv,
from="Gene",
from.source="EntrezGene",
from.org="mouse",
to.source="Ens_gene",
restricted=TRUE,
prefFilter=TRUE
)
head(res)## a b conv.from conv.to
## 1 1 0.080750138 17237 ENSMUSG00000022517
## 2 2 0.834333037 105886298 ENSMUSG00000090110
## 3 3 0.600760886 76429 ENSMUSG00000030946
## 4 4 0.157208442 80985 ENSMUSG00000027189
## 5 5 0.007399441 230514 ENSMUSG00000035212
## 6 6 0.466393497 66459 ENSMUSG00000043162Explore convertion shortest path between two identifiers
Because the conversion process takes into account several resources,
it might be useful to explore the path between two identifiers which
have been mapped. This can be achieved by the
exploreConvPath function.
The figure above shows how the ILMN_1220595 ProbeID, targeting the mouse NM_010552 transcript, can be associated to the Q16552 human protein ID in Uniprot.
Notes about converting from and to gene symbols
Canonical and non-canonical symbols are associated to genes. In some cases the same symbol (canonical or not) can be associated to several genes. This can lead to ambiguous mapping. The strategy to apply for such mapping depends on the aim of the user and his knowledge about the origin of the symbols to consider.
The complete mapping between Ensembl gene identifiers and symbols is
retrieved by using the getBeIDSymbolTable function.
compMap <- getBeIdSymbolTable(
be="Gene", source="Ens_gene", organism="rat",
restricted=FALSE
)
dim(compMap)## [1] 209352 6
head(compMap)## id symbol canonical direct preferred entity
## 1 ENSRNOG00000079114 LOC134485484 TRUE TRUE TRUE 11512563
## 2 ENSRNOG00000078531 LOC134484432 TRUE TRUE TRUE 11511580
## 3 ENSRNOG00000073047 LOC134483046 TRUE TRUE TRUE 11510323
## 4 ENSRNOG00000075384 LOC134485737 TRUE TRUE TRUE 11512792
## 5 ENSRNOG00000085489 LOC134486784 TRUE TRUE TRUE 11513789
## 6 ENSRNOG00000082375 LOC134481406 TRUE TRUE TRUE 11508810The canonical field indicates if the symbol is canonical for the identifier. The direct field indicates if the symbol is directly associated to the identifier or indirectly through a relationship with another identifier.
As an example, let’s consider the “Snca” symbol in rat. As shown below, this symbol is associated to 2 genes; it is canonical for one gene and not for another. These 2 genes are also associated to other symbols.
sncaEid <- compMap[which(compMap$symbol=="Snca"),]
sncaEid## id symbol canonical direct preferred entity
## 11576 ENSRNOG00000029408 Snca FALSE TRUE TRUE 7375332
## 61815 ENSRNOG00000008656 Snca TRUE TRUE TRUE 7344857## id symbol canonical direct preferred entity
## 11575 ENSRNOG00000029408 LOC317274 FALSE TRUE TRUE 7375332
## 11576 ENSRNOG00000029408 Snca FALSE TRUE TRUE 7375332
## 11577 ENSRNOG00000029408 Mageb16 TRUE TRUE TRUE 7375332
## 61814 ENSRNOG00000008656 MGC105443 FALSE TRUE TRUE 7344857
## 61815 ENSRNOG00000008656 Snca TRUE TRUE TRUE 7344857The getBeIdDescription function described before,
reports only one symbol for each identifier. Canonical and direct
symbols are prioritized.
getBeIdDescription(
sncaEid$id,
be="Gene", source="Ens_gene", organism="rat"
)## id symbol name preferred
## ENSRNOG00000029408 ENSRNOG00000029408 Mageb16 MAGE family member B16 TRUE
## ENSRNOG00000008656 ENSRNOG00000008656 Snca synuclein alpha TRUE
## db.version db.deprecated
## ENSRNOG00000029408 116 FALSE
## ENSRNOG00000008656 116 FALSEThe convBeIds works differently in order to provide a
mapping as exhaustive as possible. If a symbol is associated to several
input identifiers, non-canonical associations with this symbol are
removed if a canonical association exists for any other identifier. This
can lead to inconsistent results, depending on the user input, as show
below.
convBeIds(
sncaEid$id[1],
from="Gene", from.source="Ens_gene", from.org="rat",
to.source="Symbol"
)## from to to.preferred to.entity
## 3 ENSRNOG00000029408 LOC317274 NA 7375332
## 2 ENSRNOG00000029408 Mageb16 NA 7375332
## 1 ENSRNOG00000029408 Snca NA 7375332
convBeIds(
sncaEid$id[2],
from="Gene", from.source="Ens_gene", from.org="rat",
to.source="Symbol"
)## from to to.preferred to.entity
## 2 ENSRNOG00000008656 MGC105443 NA 7344857
## 1 ENSRNOG00000008656 Snca NA 7344857
convBeIds(
sncaEid$id,
from="Gene", from.source="Ens_gene", from.org="rat",
to.source="Symbol"
)## from to to.preferred to.entity
## 2 ENSRNOG00000008656 MGC105443 NA 7344857
## 1 ENSRNOG00000008656 Snca NA 7344857
## 5 ENSRNOG00000029408 LOC317274 NA 7375332
## 4 ENSRNOG00000029408 Mageb16 NA 7375332In the example above, when the query is run for each identifier independently, the association to the “Snca” symbol is reported for both. However, when running the same query with the 2 identifiers at the same time, the “Snca” symbol is reported only for one gene corresponding to the canonical association. An additional filter can be used to only keep canonical symbols:
convBeIds(
sncaEid$id,
from="Gene", from.source="Ens_gene", from.org="rat",
to.source="Symbol",
canonical=TRUE
)## from to to.preferred to.entity
## 1 ENSRNOG00000008656 Snca NA 7344857
## 2 ENSRNOG00000029408 Mageb16 NA 7375332Finally, as shown below, when running the query the other way, “Snca” is only associated to the gene for which it is the canonical symbol.
convBeIds(
"Snca",
from="Gene", from.source="Symbol", from.org="rat",
to.source="Ens_gene"
)## from to to.preferred to.entity
## 1 Snca ENSRNOG00000008656 TRUE 7344857Therefore, the user should chose the function to use with care when needing to convert from or to gene symbol.
An interactive dictionary: Shiny module
IDs, symbols and names can be seeked without knowing the original biological entity or probe. Then the results can be converted to the context of interest.
searched <- searchBeid("sv2A")
toTake <- which(searched$organism=="Homo sapiens")[1]
relIds <- geneIDsToAllScopes(
geneids=searched$GeneID[toTake],
source=searched$Gene_source[toTake],
organism=searched$organism[toTake]
)A Shiny gadget integrating these two function has been developped and is also available as an Rstudio addins.
relIds <- findBeids()It relies on a Shiny module (beidsServer() and
beidsUI() functions) made to facilitate the development of
applications focused on biological entity related information. The code
below shows a minimum example of such an application.
library(shiny)
library(BED)
library(DT)
ui <- fluidPage(
beidsUI("be"),
fluidRow(
column(
12,
tags$br(),
h3("Selected gene entities"),
DTOutput("result")
)
)
)
server <- function(input, output){
found <- beidsServer("be", toGene=TRUE, multiple=TRUE, tableHeight=250)
output$result <- renderDT({
req(found())
toRet <- found()
datatable(toRet, rownames=FALSE)
})
}
shinyApp(ui = ui, server = server)Session info
## R version 4.6.0 (2026-04-24)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.4 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=en_GB.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=en_GB.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=en_GB.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=en_GB.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Europe/Rome
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] BED_1.6.3 visNetwork_2.1.4 neo2R_3.0.0 knitr_1.51
##
## loaded via a namespace (and not attached):
## [1] miniUI_0.1.2 jsonlite_2.0.0 dplyr_1.2.1 compiler_4.6.0
## [5] promises_1.5.0 Rcpp_1.1.1-1.1 tidyselect_1.2.1 stringr_1.6.0
## [9] later_1.4.8 jquerylib_0.1.4 systemfonts_1.3.2 textshaping_1.0.5
## [13] yaml_2.3.12 fastmap_1.2.0 mime_0.13 R6_2.6.1
## [17] generics_0.1.4 curl_7.1.0 httr2_1.2.3 htmlwidgets_1.6.4
## [21] tibble_3.3.1 desc_1.4.3 shiny_1.14.0 bslib_0.11.0
## [25] pillar_1.11.1 rlang_1.2.0 DT_0.34.0 cachem_1.1.0
## [29] stringi_1.8.7 httpuv_1.6.17 xfun_0.59 fs_2.1.0
## [33] sass_0.4.10 otel_0.2.0 cli_3.6.6 withr_3.0.3
## [37] pkgdown_2.2.0 magrittr_2.0.5 digest_0.6.39 xtable_1.8-8
## [41] rstudioapi_0.19.0 rappdirs_0.3.4 lifecycle_1.0.5 vctrs_0.7.3
## [45] evaluate_1.0.5 glue_1.8.1 ragg_1.5.2 rmarkdown_2.31
## [49] tools_4.6.0 pkgconfig_2.0.3 htmltools_0.5.9